 dsb: a method for normalizing and denoising antibody derived tag data
from CITE-seq, ASAP-seq, TEA-seq and related assays.
dsb: a method for normalizing and denoising antibody derived tag data
from CITE-seq, ASAP-seq, TEA-seq and related assays.The dsb R package is available on CRAN: latest dsb
release
to install in R: install.packages('dsb')
Mulè, Martins, and Tsang, Nature Communications (2022) describes this method and deconvolution of ADT noise sources.
dsb is also available in Python through muon
Check out Recent Publications that used this method for ADT normalization.
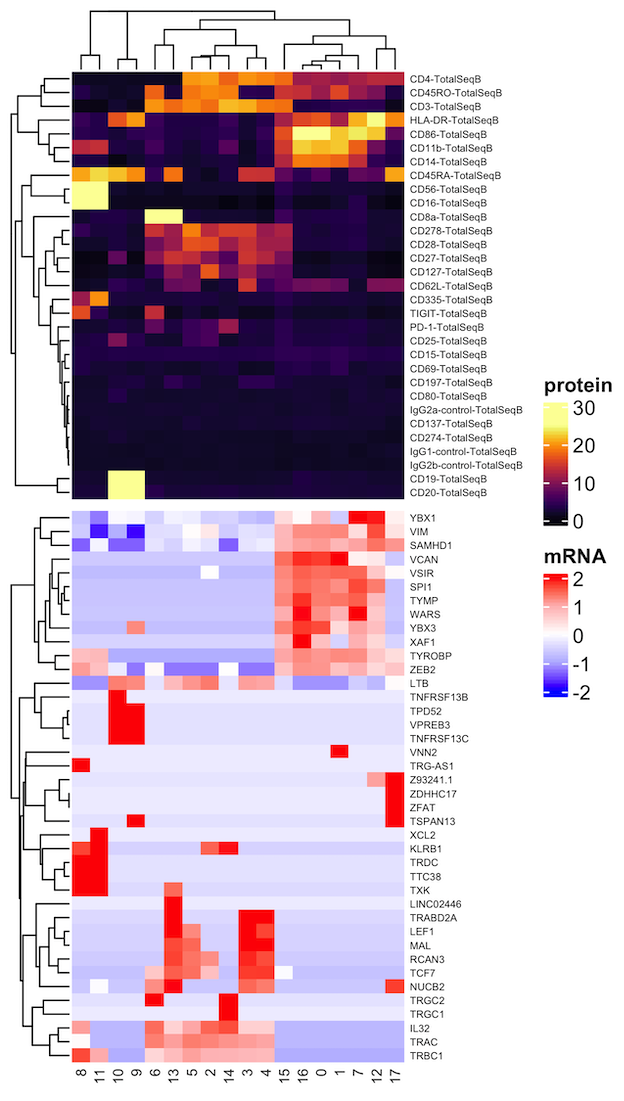
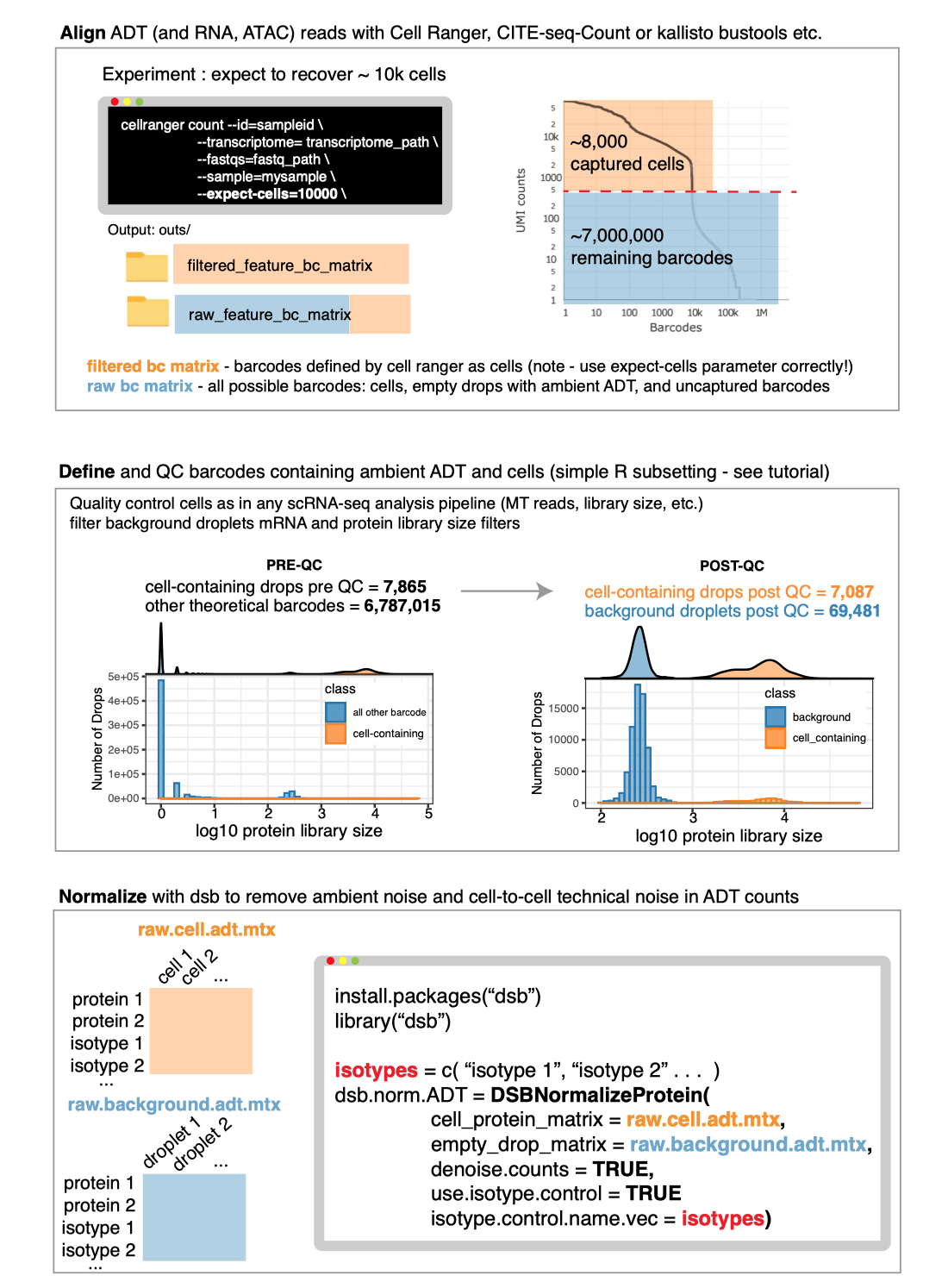
In the first Vignette, we demonstrate an end-to-end basic CITE-seq analysis starting from UMI count alignment output files from Cell Ranger. Standard output files from Cell Ranger are perfectly set up to use dsb. Our method is also compatible with any alignment tool; see: using other alignment tools. We load unfiltered UMI data containing cells and empty droplets, perform QC on cells and background droplets, normalize with dsb, and demonstrate protein-based clustering and multimodal RNA+Protein joint clustering using dsb normalized values with Seurat’s Weighted Nearest Neighbor method.
Protein data derived from sequencing antibody derived tags (ADTs) in CITE-seq and other related assays has substantial background noise. Our paper outlines experiments and analysis designed to dissect sources of noise in ADT data we used to developed our method. We found all experiments measuring ADTs capture protein-specific background noise because ADT reads in empty / background drops (outnumbering cell-containing droplets > 10-fold in all experiments) were highly concordant with ADT levels in unstained spike-in cells. We therefore utilize background droplets which capture the ambient component of protein background noise to correct values in cells. We also remove technical cell-to-cell variations by defining each cell’s dsb “technical component”, a conservative adjustment factor derived by combining isotype control levels with each cell’s specific background level fitted with a single cell model.
The method is carried out in a single step with a call to the
DSBNormalizeProtein() function.
cells_citeseq_mtx - a raw ADT count matrix
empty_drop_citeseq_mtx - a raw ADT count matrix from
non-cell containing empty / background droplets.
denoise.counts = TRUE - implement step II to define and
remove the ‘technical component’ of each cell’s protein library.
use.isotype.control = TRUE - include isotype controls in
the modeled dsb technical component.
# install.packages('dsb')
library(dsb)
adt_norm = DSBNormalizeProtein(
cell_protein_matrix = cells_citeseq_mtx,
empty_drop_matrix = empty_drop_citeseq_mtx,
denoise.counts = TRUE,
use.isotype.control = TRUE,
isotype.control.name.vec = rownames(cells_citeseq_mtx)[67:70]
)For a full tutorial vignette using 10X genomics data demonstrating how to quality control cells and empty droplets, normalize ADTs with dsb and ue these values with joint mRNA and protein clustering to generate the joint map below, please see the main vignette on CRAN
Publications from outside institutes
Jardine et
al. Nature (2021)
Mimitou et.
al Nature Biotechnology (2021)
COMBAT
consortium et al. Cell (2022)
Publications from the Tsang lab
Kotliarov et
al. Nature Medicine (2020)
Liu et
al. Cell (2021)
dsb was developed prior to 10X Genomics supporting CITE-seq or hashing data and we routinely use other alignment pipelines.
A note on alignment and how to use dsb with Cell Ranger is detailed in the main vignette. Cells and empty droplets are used by default by dsb.
To use dsb properly with CITE-seq-Count you need to align background.
One way to do this is to set the -cells argument to ~
200000. That will align the top 200000 barcodes in terms of ADT library
size, making sure you capture the background. Please refer to CITE-seq-count
documentation
CITE-seq-Count -R1 TAGS_R1.fastq.gz -R2 TAGS_R2.fastq.gz \
-t TAG_LIST.csv -cbf X1 -cbl X2 -umif Y1 -umil Y2 \
-cells 200000 -o OUTFOLDERIf you already aligned your mRNA with Cell Ranger or something else but wish to use a different tool like kallisto or Cite-seq-count for ADT alignment, you can provide the latter with whitelist of cell barcodes to align. A simple way to do this is to extract all barcodes with at least k mRNA where we set k to a tiny number to retain cells and cells capturing ambient ADT reads:
library(Seurat)
umi = Read10X(data.dir = 'data/raw_feature_bc_matrix/')
k = 3
barcode.whitelist =
rownames(
CreateSeuratObject(counts = umi,
min.features = k, # retain all barcodes with at least k raw mRNA
min.cells = 800, # this just speeds up the function by removing genes.
)@meta.data
)
write.table(barcode.whitelist,
file =paste0(your_save_path,"barcode.whitelist.tsv"),
sep = '\t', quote = FALSE, col.names = FALSE, row.names = FALSE)With the example dataset in the vignette this retains about 150,000 barcodes.
Now you can provide that as an argument to -wl in
CITE-seq-count to align the ADTs and then proceed with the dsb analysis
example.
CITE-seq-Count -R1 TAGS_R1.fastq.gz -R2 TAGS_R2.fastq.gz \
-t TAG_LIST.csv -cbf X1 -cbl X2 -umif Y1 -umil Y2 \
-wl path_to_barcode.whitelist.tsv -o OUTFOLDERThis whitelist can also be provided to Kallisto.
kallisto
bustools documentation
kb count -i index_file -g gtf_file.t2g -x 10xv3 \
-t n_cores -w path_to_barcode.whitelist.tsv -o output_dir \
input.R1.fastq.gz input.R2.fastq.gzNext one can similarly define cells and background droplets empirically with protein and mRNA based thresholding as outlined in the main tutorial.
Note whether or not you use dsb, if you want to define cells
using the filtered_feature_bc_matrix file, you should make
sure to properly set the Cell Ranger --expect-cells
argument roughly equal to the estimated cell recovery per lane based on
number of cells you loaded in the experiment. see the
note from 10X about this. The default value of 3000 is relatively
low for modern experiments. Note cells and empty droplets can also be
defined directly from the raw_feature_bc_matrix using any
method, including simple protein and mRNA library size based
thresholding because this contains all droplets.
Topics covered in other vignettes on CRAN: Integrating dsb with Bioconductor, integrating dsb with python/Scanpy, Using dsb with data lacking isotype controls, integrating dsb with sample multiplexing experiments, using dsb on data with multiple batches, advanced usage - using a different scale / standardization based on empty droplet levels, returning internal stats used by dsb, outlier clipping with the quantile.clipping argument, other FAQ.